Contrasts and Post-Hoc Testing
After fitting statistical models like ANOVA, linear regression, GLMs, or mixed-effects models, we often want to ask more specific questions than the overall model summary provides. For instance, a significant F-test in an ANOVA tells us that at least one group differs from another, but it doesn’t tell us which specific groups differ. Similarly, regression coefficients tell us about the effect of a predictor compared to a reference level, but we might be interested in comparing other levels or combinations of levels directly. This is where contrasts and post-hoc testing become essential.
Introduction
Contrasts are essentially planned comparisons between specific means or combinations of means within our model. They allow us to test precise hypotheses derived from our research questions. For example, if we have three treatment groups (Control, Treatment A, Treatment B), we might want to compare the average outcome of the two treatment groups against the control, or compare Treatment A directly against Treatment B. Post-hoc tests are often performed after an initial significant result (like an ANOVA F-test or an assessment of the model summary) to explore which specific group differences are driving that significance, often involving multiple pairwise comparisons.
While it’s possible to calculate contrasts manually, this can become complex, especially with interaction terms or complicated models like mixed-effects models. Fortunately, the R ecosystem provides powerful tools to simplify this process. The emmeans package (Estimated Marginal MEanS) is particularly useful. It allows us to easily compute estimated marginal means (also known as least-squares means), perform various contrasts, conduct post-hoc tests, and visualize results, all while properly accounting for the model structure and potential covariates. This tutorial will introduce the concept of contrasts and demonstrate how to use the emmeans package in R to perform insightful post-hoc analyses.
Example 1: Plant Defenses in Kluane – Using emmeans
To illustrate how contrasts work, we’ll use data from the Kluane Boreal Forest Ecosystem Project. This long-term experiment investigated how fertilization and the exclusion of herbivores affect vegetation dynamics. The specific data we’ll analyze concerns the concentration of phenolic compounds (a measure of plant defense) in yarrow (Achillea millefolium).
The experiment involved sixteen 5x5m plots. Each plot was assigned one of four treatments: control (no manipulation), exclosure (fenced to keep herbivores out), fertilizer (N-P-K added), or both (fenced and fertilized). Each plot was then split in half. One half received its assigned treatment continuously for 20 years (“permanent” duration). The other half received the treatment for the first 10 years, after which the treatment was stopped, allowing the vegetation to revert towards its untreated state (“reverse” duration). Our goal is to understand how these treatments and their duration influenced yarrow’s phenolic concentrations.
Data Preparation and Visualization
First, let’s load the necessary libraries and the data. We’ll convert plot, treatment, and duration into factors. Notice that we use fct_relevel to set “control” as the reference level for the treatment factor, which influences how R encodes the model later.
library(tidyverse)
library(lmerTest)
library(broom.mixed) # For tidying model outputs, especially with emmeans
library(emmeans) # For calculating estimated marginal means and contrasts
# Load the data
df_kluane <- read_csv("https://www.zoology.ubc.ca/~bio501/R/data/kluane.csv") %>%
mutate(
plot = as.factor(plot),
# Set reference level to 'control'
treatment = as.factor(treatment) %>% fct_relevel("control", "exclosure", "fertilizer", "both"),
duration = as.factor(duration) # Levels: permanent, reverse
)
# Check the structure
str(df_kluane)tibble [32 × 4] (S3: tbl_df/tbl/data.frame)
$ plot : Factor w/ 16 levels "1","2","3","4",..: 1 1 2 2 3 3 4 4 5 5 ...
$ treatment: Factor w/ 4 levels "control","exclosure",..: 1 1 3 3 1 1 3 3 4 4 ...
$ duration : Factor w/ 2 levels "permanent","reverse": 1 2 1 2 1 2 1 2 1 2 ...
$ phen.ach : num [1:32] 44 36.5 14.2 28.6 42.4 ...Remember your training! It’s always wise to visualize the data. We’ll plot the log-transformed phenolic concentrations, faceted by treatment, showing the change between the “permanent” and “reverse” durations. Lines connect the two measurements from the same plot.
df_kluane %>%
ggplot(aes(y = log(phen.ach), x = duration, fill = duration, color = duration)) +
geom_line(aes(group = plot), col = "black", alpha=0.3) + # Connect points within plots
geom_point(size = 3, position = position_dodge(width = 0.2)) + # Dodge points slightly
facet_wrap(~treatment, nrow = 1) +
labs(
title = "Yarrow Phenolic Concentration by Treatment and Duration",
x = "Treatment Duration",
y = "Log Phenolics Concentration (mg TAE/g DW)",
color = "Duration", fill = "Duration"
) +
ggthemes::theme_base()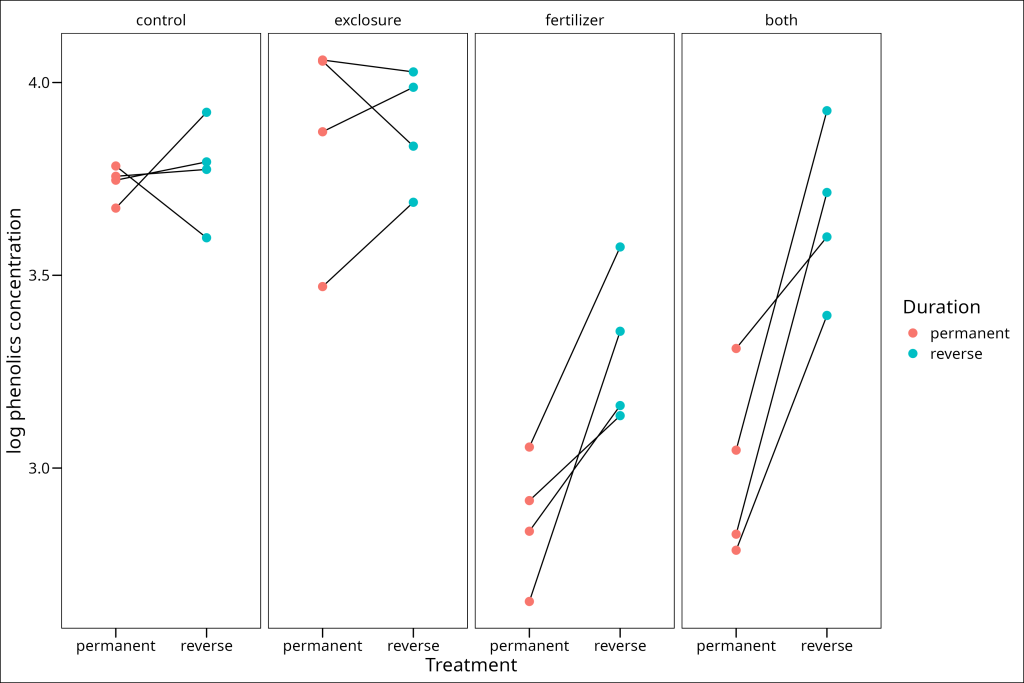
The plot suggests potential interactions. For instance, in the fertilizer and “both” treatments, phenolic concentrations seem to increase when the treatment stops (permanent –> reverse), while this pattern is less clear or absent in the control and exclosure treatments.
Fitting the Mixed-Effects Model
We expect measurements from the same plot to be correlated. Therefore, we’ll fit a linear mixed-effects model using lmer from the lmerTest package. We model log-transformed phenolics (log(phen.ach)) as a function of the interaction between treatment and duration, including plot as a random intercept to account for baseline differences between plots.
model_kluane <- lmer(
log(phen.ach) ~ treatment * duration + (1 | plot),
data = df_kluane
)
# Display the model summary
summary(model_kluane)Linear mixed model fit by REML. t-tests use Satterthwaite's method ['lmerModLmerTest']
Formula: log(phen.ach) ~ treatment * duration + (1 | plot)
Data: df_kluane
REML criterion at convergence: -1.4
Scaled residuals:
Min 1Q Median 3Q Max
-1.55278 -0.58471 0.03425 0.52871 1.61347
Random effects:
Groups Name Variance Std.Dev.
plot (Intercept) 0.01292 0.1137
Residual 0.02413 0.1553
Number of obs: 32, groups: plot, 16
Fixed effects:
Estimate Std. Error df t value Pr(>|t|)
(Intercept) 3.74031 0.09624 21.39871 38.865 < 2e-16 ***
treatmentexclosure 0.12388 0.13610 21.39871 0.910 0.37284
treatmentfertilizer -0.87526 0.13610 21.39871 -6.431 2.06e-06 ***
treatmentboth -0.74732 0.13610 21.39871 -5.491 1.78e-05 ***
durationreverse 0.03190 0.10984 12.00000 0.290 0.77648
treatmentexclosure:durationreverse -0.01122 0.15534 12.00000 -0.072 0.94362
treatmentfertilizer:durationreverse 0.40963 0.15534 12.00000 2.637 0.02169 *
treatmentboth:durationreverse 0.63438 0.15534 12.00000 4.084 0.00152 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Correlation of Fixed Effects:
(Intr) trtmntx trtmntf trtmntb drtnrv trtmntx: trtmntf:
trtmntxclsr -0.707
trtmntfrtlz -0.707 0.500
treatmntbth -0.707 0.500 0.500
duratinrvrs -0.571 0.404 0.404 0.404
trtmntxcls: 0.404 -0.571 -0.285 -0.285 -0.707
trtmntfrtl: 0.404 -0.285 -0.571 -0.285 -0.707 0.500
trtmntbth:d 0.404 -0.285 -0.285 -0.571 -0.707 0.500 0.500 Interpreting the Default Summary (and its Limitations)
The model summary provides estimates for the fixed effects. Understanding these requires knowing how R handles categorical predictors. By default, R uses “treatment contrasts” (confusingly named, unrelated to the contrasts we’ll discuss later). With treatment contrasts, the first level of a factor (which we set to “control” for treatment and which is “permanent” for duration by default) becomes the reference level.
Here’s how to interpret the coefficients:
- (Intercept): 3.740 is the estimated mean log phenolic concentration for the reference group:
treatment="control"andduration="permanent".
- treatmentexclosure: 0.124 is the difference in mean log phenolics between the
exclosuretreatment and thecontroltreatment, specifically whenduration="permanent".
- treatmentfertilizer: -0.875 is the difference between
fertilizerandcontrolwhenduration="permanent".
- treatmentboth: -0.747 is the difference between
bothandcontrolwhenduration="permanent".
- durationreverse: 0.032 is the difference between
duration="reverse"andduration="permanent", specifically for thecontroltreatment.
- treatmentexclosure:durationreverse: -0.011 is an interaction term. It represents how much the effect of switching from permanent to reverse duration *differs* in the
exclosuretreatment compared to thecontroltreatment. In other words, it’s (Exclosure:Reverse – Exclosure:Permanent) – (Control:Reverse – Control:Permanent).
- treatmentfertilizer:durationreverse: 0.410 is an interaction term comparing the duration effect in the
fertilizertreatment to the duration effect in thecontroltreatment.
- treatmentboth:durationreverse: 0.634 is the interaction term comparing the duration effect in the
bothtreatment to the duration effect in thecontroltreatment.
While informative, this summary can’t directly answer many biologically relevant questions. For example:
1. What is the estimated mean phenolic concentration for the fertilizer group under the reverse duration? (We only get the difference relative to control permanent).
2. Is the mean phenolic concentration in the “both” treatment under the reverse duration significantly different from the fertilizer treatment under the reverse duration?
3. Is the effect of switching from permanent to reverse duration significantly different between the “fertilizer” and “both” treatments? (The summary only gives comparisons relative to control).
4. Is the average phenolic concentration for a specific group (e.g., fertilizer-reverse) significantly different from a specific hypothesized value (e.g., 3.0)?
To answer these kinds of targeted questions, we need to perform specific comparisons or contrasts, which is where the emmeans package comes in.
Using emmeans for Specific Comparisons
The emmeans package calculates “Estimated Marginal Means” (EMMs), which are essentially model-predicted means for different groups or combinations of factor levels, averaged over any covariates. It provides a flexible framework for performing various types of contrasts based on these EMMs.
1. One-Sample Comparisons: Testing Against a Specific Value
Sometimes, we want to test if the mean of a specific group differs from a known or hypothesized value. Let’s address Question 4 from before: Is the mean log phenolic concentration of the fertilizer plots under the ‘reverse’ duration significantly greater than 3?
We use the emmeans() function, specifying the model and the factors involved (~ treatment * duration). The at argument filters the results to only the combination we care about (treatment = "fertilizer" and duration = "reverse"). We set our null hypothesis value with null = 3 and specify a one-sided test (side = "right") because we’re interested if the concentration is *greater* than 3. Finally, broom.mixed::tidy() cleans up the output to include p-values of interest.
# Q: Is fertilizer-reverse > 3?
emmeans(
model_kluane, # The fitted model
~ treatment * duration, # Specify factors involved
at = list(treatment = c("fertilizer"), duration = "reverse"), # Filter to the specific group of interest
null = 3, # Null hypothesis value
adjust = "none", # No adjustment needed for a single test
side = "right" # One-sided test (greater than)
) %>%
broom.mixed::tidy() # Tidy the output# A tibble: 1 × 8
treatment duration estimate std.error df null statistic p.value
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 fertilizer reverse 3.31 0.0962 21.4 3 3.19 0.00219The output shows an estimated mean log phenolic concentration of 3.31 for this group. The p-value (0.00219) is less than the conventional alpha level of 0.05, suggesting strong evidence against the null hypothesis. We can conclude that the mean log phenolic concentration for fertilizer plots under the reverse duration is significantly greater than 3.
2. Two-Sample Comparisons: Comparing Specific Groups
A more common task is comparing the means of two specific groups. Let’s address Question 2: Is there a significant difference between the control and fertilized plots, specifically under the ‘reverse’ duration condition?
First, we calculate the EMMs for the groups we want to compare using emmeans(), filtering with at. When conducting two-sample comparisons, we need to pass the initial emmeans() output into the emmeans::contrast() function which takes care of performing the contrast comparison. Within this function, we specify method = "pairwise" to compare all pairs within the filtered groups (in this case, just control-reverse vs. fertilizer-reverse). Since we are performing a comparison that wasn’t necessarily planned a priori based on the overall model significance, it’s good practice to adjust the p-value for multiple comparisons with the adjust argument, even if there’s only one pair here.
emmeans(
model_kluane,
~ duration * treatment, # Order matters slightly for output formatting
# Filter to the two groups to compare
at = list(treatment = c("control", "fertilizer"), duration = c("reverse"))
) %>%
emmeans::contrast(
method = "pairwise", # Compare pairs of the selected EMMs
adjust = "bonferroni" # Adjust p-value
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term contrast null.value estimate std.error df statistic p.value
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 duration*treatment reverse control - reverse fertilizer 0 0.466 0.136 21.4 3.42 0.00252The contrast compares “reverse control” minus “reverse fertilizer”. The estimate of this difference is 0.466. The p-value (0.00252) indicates a significant difference between these two specific group means, after Bonferroni adjustment.
The method argument
The method argument in emmeans::contrast() (and similarly the values used within the interaction argument) defines the type of comparisons you want to make. Here are some common choices:
"pairwise": Compares all pairs of means within the specified groups. This is often used for post-hoc exploration after a significant overall effect. Example: Comparing Control vs TreatmentA, Control vs TreatmentB, and TreatmentA vs TreatmentB."trt.vs.ctrl": Compares each specified group mean against the first group (often the control or reference level). Example: Comparing TreatmentA vs Control and TreatmentB vs Control."consec": Compares adjacent levels of a factor. Useful if the factor levels have a natural ordering. Example: Comparing Level1 vs Level2, Level2 vs Level3, etc.
When using the interaction argument (e.g., interaction = "pairwise" or interaction = c("pairwise", "trt.vs.ctrl")), these same methods are applied to the differences between levels of interacting factors, allowing you to test how effects change across conditions.
3. Interaction Contrasts: Comparing Differences of Differences
Interaction contrasts allow us to test if the effect of one factor differs across the levels of another factor. Let’s tackle Question 3: We saw visually that phenolics seemed to increase from ‘permanent’ to ‘reverse’ duration in both the fertilizer and ‘both’ treatments. Is this increase (the duration effect) significantly more pronounced in the ‘both’ treatment compared to the ‘fertilizer’ treatment?
Again, we start with emmeans(), specifying the factors involved and filtering using at to include only the levels relevant to our question (treatment = c("fertilizer", "both"); duration levels are implicitly included). Because we a interested in differences between two or more groups, we need to pipe into emmeans::contrast(). As we wish the test for simultaneous differences between these two variables (difference of differences), we use the argument interaction = "pairwise" instead of method = "pairwise". This tests if the pairwise difference between duration levels (reverse – permanent) is itself different between the treatment levels (both – fertilizer). Essentially, it calculates: (Both:Reverse – Both:Permanent) – (Fertilizer:Reverse – Fertilizer:Permanent).
emmeans(
model_kluane,
~ treatment * duration,
at = list(treatment = c("fertilizer", "both")) # Focus on these treatments
) %>%
contrast(
interaction = "pairwise", # Test the difference of differences
adjust = "bonferroni"
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term treatment_trt.vs.ctrl duration_trt.vs.ctrl estimate std.error df statistic p.value
<chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 treatment*duration both - fertilizer reverse - permanent 0.225 0.155 12.0 1.45 0.174The output explicitly shows the interaction being tested: `both – fertilizer` difference within the `reverse – permanent` difference. The estimated difference in the duration effect is 0.225. However, the p-value (0.174) is not statistically significant. This suggests that while both treatments showed an increase in phenolics when the treatment stopped, the magnitude of this increase was not significantly different between the ‘both’ and ‘fertilizer’ treatments.
Connecting Back to Model Coefficients
It’s useful to see how emmeans relates to the original model coefficients. Recall the interaction coefficient treatmentboth:durationreverse (0.634, p=0.00152) from the summary(model_kluane) output. This coefficient represented the interaction comparing the duration effect (reverse vs. permanent) in the ‘both’ treatment relative to the control treatment. Let’s replicate this specific comparison using emmeans:
emmeans(
model_kluane,
~ treatment * duration,
# Focus on control and both treatments, and both durations
at = list(treatment = c("control", "both"))
) %>%
contrast(
interaction = "pairwise", # Compare duration effect (rev-perm) between treatments (both-ctrl)
adjust = "bonferroni"
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term treatment_pairwise duration_pairwise estimate std.error df statistic p.value
<chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 treatment*duration both - control reverse - permanent 0.634 0.155 12 4.08 0.00152Et voila, the estimate (0.634) and p-value (0.00152) perfectly match the corresponding interaction coefficient from the model summary. This demonstrates that while the default summary only provides comparisons relative to the reference levels, emmeans allows us to calculate any desired comparison, including those implicitly shown in the summary and many others that are not.
Example 2: Analyzing Slopes with emtrends
While emmeans is excellent for comparing estimated marginal means (i.e., adjusted group averages), sometimes our hypotheses concern the slopes of relationships. For instance, we might want to know if the rate of change of a response variable with respect to a continuous predictor differs between groups. This is where the emtrends() function from the emmeans package comes in handy. It estimates and compares the slopes of fitted lines for different factor levels.
To illustrate emtrends, we will use a fabricated dataset. Imagine a study where we measure the height and weight of individuals belonging to three different arbitrary groups (A, B, C) and two sexes (Male, Female). We are interested in how the relationship between height (predictor) and weight (response) changes across these groups and sexes. The data has been deliberately constructed with different height-weight relationships, including a specific interaction making the slope steeper for males in Group C.
Data Generation and Visualization
Let us first generate this dataset and visualize the relationship between height and weight for each group and sex combination.
# Set seed for reproducibility
set.seed(123)
# Create the dataset
n_per_group <- 30 # 30 individuals per group
n_groups <- 3 # 3 groups (A, B, C)
# Generate the data
df_hws <- tibble(
Group = rep(c("A", "B", "C"), each = n_per_group) %>% fct_relevel("A", "B", "C"),
Sex = rep(c("Male", "Female"), each = n_per_group / 2, times = n_groups) %>% fct_relevel("Female", "Male"),
Height = case_when(
Sex == "Male" ~ rnorm(n_per_group * n_groups, mean = 180, sd = 10),
Sex == "Female" ~ rnorm(n_per_group * n_groups, mean = 165, sd = 8)
),
Weight = case_when(
# Base weight (males heavier than females)
Sex == "Male" ~ rnorm(n_per_group * n_groups, mean = 80, sd = 10),
Sex == "Female" ~ rnorm(n_per_group * n_groups, mean = 65, sd = 8)
) +
# Add height effect (different slopes for each group)
case_when(
Group == "A" ~ 0.5 * (Height - mean(Height)),
Group == "B" ~ 0.7 * (Height - mean(Height)),
Group == "C" ~ 1.2 * (Height - mean(Height))
) +
# Add interaction effect for Group C males
case_when(
Group == "C" & Sex == "Male" ~ 1.5 * (Height - mean(Height)),
TRUE ~ 0
) +
# Add some statistical noise
rnorm(n_per_group * n_groups, mean = 0, sd = 5)
)
str(df_hws)tibble [90 × 4] (S3: tbl_df/tbl/data.frame)
$ Group : Factor w/ 3 levels "A","B","C": 1 1 1 1 1 1 1 1 1 1 ...
$ Sex : Factor w/ 2 levels "Female","Male": 2 2 2 2 2 2 2 2 2 2 ...
$ Height: num [1:90] 174 178 196 181 181 ...
$ Weight: num [1:90] 69.3 98.5 89.5 80.6 86.2 ...# Plot the data
df_hws %>%
ggplot(aes(x = Height, y = Weight, color = Sex, fill = Sex)) +
geom_point(alpha = 0.5) +
geom_smooth(method = "lm", se = TRUE) +
facet_wrap(~Group) +
labs(
title = "Height vs. Weight by Group and Sex",
x = "Height (cm)",
y = "Weight (kg)"
) +
ggthemes::theme_base()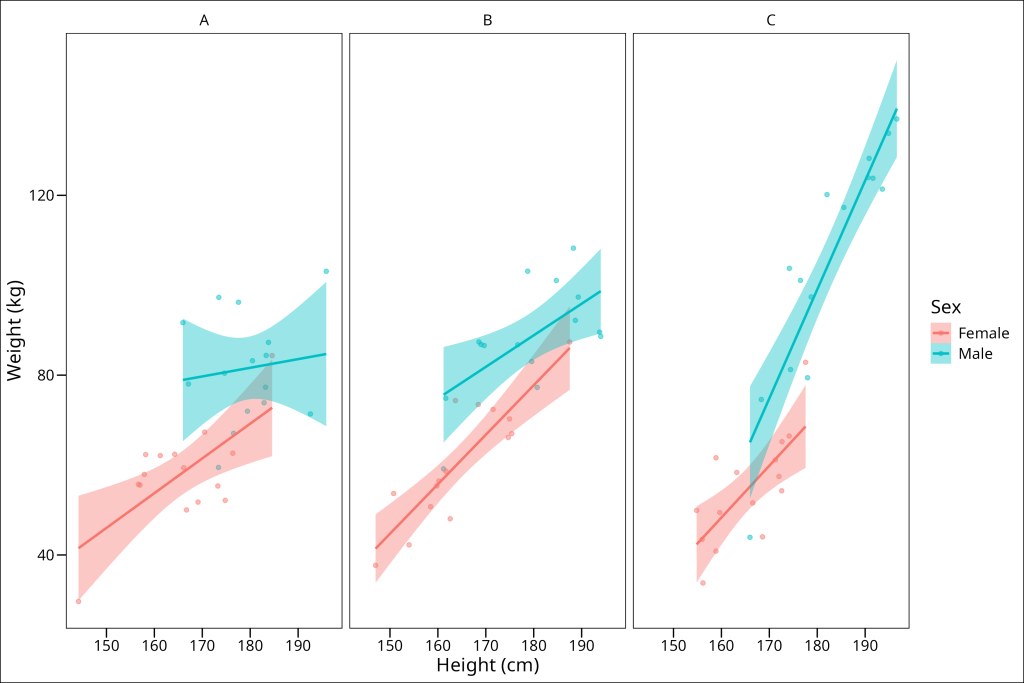
The plot shows varying relationships between height and weight across the groups and sexes. As intended by the data generation, the slope for Group C Males appears noticeably steeper than for other groups.
Fitting the Linear Model
We fit a linear model (lm) predicting Weight based on Group, Sex, and Height, including all possible interactions (Group * Sex * Height). This allows the relationship (slope) between Height and Weight to differ for every combination of Group and Sex.
model_hws <- lm(Weight ~ Group * Sex * Height, data = df_hws)
summary(model_hws)Call:
lm(formula = Weight ~ Group * Sex * Height, data = df_hws)
Residuals:
Min 1Q Median 3Q Max
-21.1212 -6.0448 0.0515 5.6366 18.8071
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -69.9453 43.1485 -1.621 0.10905
GroupB -50.5078 58.8345 -0.858 0.39326
GroupC -66.3386 70.8607 -0.936 0.35207
SexMale 116.8949 71.4718 1.636 0.10597
Height 0.7731 0.2604 2.968 0.00398 **
GroupB:SexMale -34.0398 92.3319 -0.369 0.71337
GroupC:SexMale -317.5395 102.8442 -3.088 0.00279 **
GroupB:Height 0.3281 0.3558 0.922 0.35922
GroupC:Height 0.3807 0.4277 0.890 0.37612
SexMale:Height -0.5803 0.4106 -1.414 0.16148
GroupB:SexMale:Height 0.1817 0.5328 0.341 0.73394
GroupC:SexMale:Height 1.8487 0.5938 3.113 0.00259 **
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 9.758 on 78 degrees of freedom
Multiple R-squared: 0.8552, Adjusted R-squared: 0.8348
F-statistic: 41.88 on 11 and 78 DF, p-value: < 2.2e-16The model summary shows significant two-way (GroupC:SexMale) and three-way (GroupC:SexMale:Height) interactions involving Group C and Males. This strongly suggests the relationship between height and weight is different for males in group C. The emtrends function provides a more direct way to test specific hypotheses about these slopes.
1. One-Sample Slope Test
Question: Do males in Group A have a significantly positive relationship (i.e., slope > 0) between height and weight?
We use emmeans::emtrends(), which fortunately behaves almost identically to its emmeans::emmeans() counterpart – specifying the model, the factors defining the groups (~ Group * Sex). The at argument filters to “Male” in “Group A”. We test against a null hypothesis of 0 (null = 0) using a one-sided test (side = "right"). Crucially, this function has one more important argument var, which is the the continuous variable for which we want to compare the slopes for. In our case, "Height".
emmeans::emtrends(
model_hws,
~ Group * Sex,
at = list(Group = "A", Sex = "Male"),
var = "Height",
side = "right",
null = 0
) %>%
broom.mixed::tidy()# A tibble: 1 × 7
Group Sex Height.trend std.error df statistic p.value
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 A Male 0.193 0.317 78 0.607 0.273The estimated slope (Height.trend) for Group A males is 0.193. The p-value of 0.273 suggests that this slope is not significantly greater than zero. We do not have sufficient evidence to conclude there is a positive relationship between height and weight for males in Group A in this dataset.
2. Two-Sample Slope Comparison
Question: Is there a difference in the relationship (slope) between height and weight for males versus females within Group C?
We first get the trends for Males and Females in Group C using emmeans::emtrends() and the at argument, then pipe to emmeans::contrast(method = "pairwise") to compare these two slopes.
# Q: Is Height-Weight slope different between Group C Males and Group C Females?
emmeans::emtrends(
model_hws,
~ Group * Sex,
at = list(Group = "C", Sex = c("Male", "Female")),
var = "Height"
) %>%
emmeans::contrast(
method = "pairwise",
adjust = "bonferroni"
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term contrast null.value estimate std.error df statistic p.value
<chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Group*Sex C Male - C Female 0 1.27 0.429 78 2.96 0.00411The estimated difference in slopes (Male – Female in Group C) is 1.27. The very small p-value (0.00411) indicates a highly significant difference. As we deliberately designed for this dataset, males in Group C have a significantly steeper positive relationship between height and weight compared to females in Group C.
3. Interaction Test for Slopes
Question: Does the difference in height-weight slopes between males and females change significantly when moving from Group B to Group C?
We get the trends for both sexes in Groups B and C using emmeans::emtrends() and the at argument, then use emmeans::contrast(interaction = "pairwise"). This tests if the (Male slope – Female slope) difference is different between Group B and Group C.
# Q: Does (Male slope - Female slope) differ between Group B and Group C?
emmeans::emtrends(
model_hws,
~ Group * Sex,
at = list(Group = c("B", "C"), Sex = c("Male", "Female")),
var = "Height"
) %>%
emmeans::contrast(
interaction = "pairwise",
adjust = "bonferroni"
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term Group_pairwise Sex_pairwise estimate std.error df statistic p.value
<chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Group*Sex B - C Male - Female -1.67 0.547 78 -3.05 0.00315The output shows the interaction contrast: (B Male – B Female) – (C Male – C Female). The estimate is -1.67, and the p-value (0.00315) is significant. This indicates that the way the height-weight slope differs between sexes is significantly different when comparing Group B to Group C. Specifically, the male-female slope difference is much larger in Group C than in Group B.
Connecting Back to Model Coefficients
Just like with emmeans::emmeans(), we can relate emmeans::emtrends() contrasts back to the model coefficients, although it requires careful consideration of reference levels. Let us try to replicate the GroupB:SexMale:Height coefficient (Estimate = 0.1817, p = 0.734) from the summary(model_hws) output. This coefficient represents how the slope difference between male and the reference female changes when moving from the reference Group A to Group B.
We can test this using an interaction contrast focusing on Groups A and B, and both Sexes. Note the use of interaction = "trt.vs.ctrl" which compares each factor’s levels against its reference level.
# Q: Replicate the model coefficient for GroupB:SexMale:Height interaction
# This represents (B:Male - B:Female) - (A:Male - A:Female) slope differences,
# or equivalently, (B:Male - A:Male) - (B:Female - A:Female)
emmeans::emtrends(
model_hws,
~ Group * Sex,
at = list(Group = c("A", "B"), Sex = c("Male", "Female")),
var = "Height"
) %>%
emmeans::contrast(
# Compare Group B vs A (ref) within Male vs Female (ref)
interaction = c("trt.vs.ctrl", "trt.vs.ctrl"),
adjust = "none" # No adjustment for single coefficient replication
) %>%
broom.mixed::tidy()# A tibble: 1 × 8
term Group_trt.vs.ctrl Sex_trt.vs.ctrl estimate std.error df statistic p.value
<chr> <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Group*Sex B Male 0.182 0.533 78 0.341 0.734The estimate (0.182) and p-value (0.734) match the GroupB:SexMale:Height coefficient in the model summary. This again highlights that emmeans::emtrends allows flexible testing of slope comparisons, including those represented by complex interaction terms in the original model summary.
Conclusion
Contrasts and post-hoc tests are indispensable tools for moving beyond the general output of statistical models to answer specific, nuanced research questions. The emmeans package in R, with its emmeans() and emtrends() functions, provides a powerful and flexible framework for conducting these detailed analyses. Whether you are comparing adjusted group means or the slopes of relationships, emmeans simplifies the process of specifying comparisons, performing tests, and obtaining clear, interpretable results, even for complex models involving interactions and random effects.
End of Module
Great job on working through the core statistical methods in R! You’re now equipped to test hypotheses and model data. The journey doesn’t end here, however. The next module, “Ecology and Evolutionary Biology (EEB) Analyses,” takes a deeper dive into using R and specialized third-party packages for the kinds of sophisticated analyses frequently employed in EEB. Get ready to tackle some of the cornerstone analytical techniques in the field!
Whenever you’re ready, click the link below to proceed to the next module: